|
||||
|
|
ЛЕКЦИЯ № 13. Электрохимическая кинетика 1. Основные кинетические характеристики и методы их расчетов i0 – ток обмена – кинетическая характеристика равновесия между электродом и раствором при равновесном значении электродного потенциала. Токи обмена относят к 1 см2 поверхности раздела электрод-раствор. ?– коэффициент переноса заряда – характеризует степень влияния электрического поля электрода на энергию активации электрохимической стадии и определяет симметрию катодного и анодного процессов, зависит от формы потенциальных кривых. ?? 0,5. При одном и том же отклонении потенциала электрода от равновесного значения скорости реакции результативная плотность тока будет тем больше, чем выше i0. Ток обмена i0 зависит от природы электрохимической реакции, материала электрода и состава раствора. Константа скорости – скорость реакции при единичных концентрациях. Скорость прямой реакции: 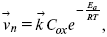 где k – константа, зависящая от свойств системы и способа выражения скорости процесса; Cox – концентрация реагирующих частиц; Ea – энергия активации разряда в отсутствии скачка потенциала между металлом и раствором. Скорость обратной реакции 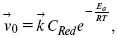 где CRed – концентрация частиц Red (восстановление продуктов); Еa– энергия активации реакции ионизации при скачке потенциала между металлом и раствором, равным нулю. Энергия активации электрохимического процесса зависит от величины электродного потенциала, природы ее непосредственных участников и электрода. Энергия активации при постоянном перенапряжении ? – эффективная энергия активации. Если энергия активации не зависит от перенапряжения, то ее появление замедляет диффузию. Метод расчета величин ?и i0 основан на явлении редоксикинетического эффекта заключается в том, что при наложении переменного тока на электрод его потенциал смещается в ту или иную сторону на некоторую величину от первоначального значения. Это смещение – редоксикинетический потенциал ?. Связь редоксикинетического потенциала ? с кинетическими параметрами а и i0 такая: если наложить переменный ток на электрод, находящийся в равновесии с соответствующими ионами в растворе, то за время катодного полупериода он окажется заполяризованным катодно, причем зависимость между ?и iпри условии замедленности стадии разряда будет передаваться уравнением 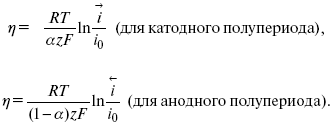 При достаточном удалении от состояния равновесия 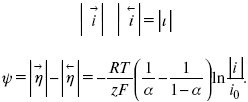 Из последнего выражения следует, если ?= 0,5, то ? = 0, чем сильнее ?отклонена от 0,5, тем больше ?. Энергия активизации – энергия, представляющая собой минимальную энергию, достаточную для осуществления акта химической реакции. 2. Уравнения электрохимической кинетики, пределы их применимости 1-й закон Фарадея устанавливает прямую пропорциональность между количеством прошедшего через систему электричества и количеством прореагировавшего вещества. ?m = kэJt = kэq, (1) где ?m – количество прореагировавшего вещества; k – коэффициент пропорциональности; q – количество электричества, равное произведению силы тока I на время t. Если q = Jt = 1, то ?m = kэ – количество вещества, прореагировавшего в результате протекания единицы количества электричества. kэ – электрохимический эквивалент. 2-й закон Фарадея устанавливает связь между количеством прореагировавшего вещества при пропускании данного количества электричества и его природой. По этому закону, при постоянном количестве прошедшего электричества массы прореагировавших веществ относятся между собой, как их химические эквиваленты А: 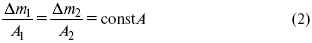 Если количество электричества равно F, числу Фарадея, то ?m1 = Fkэ1 = A1, Fkэ при q = 1F, то 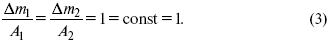 Уравнение (3) позволяет объединить оба закона Фарадея в виде одного общего закона, по которому количество электричества (1F = 96500k) всегда изменяет электрохимически массу любого вещества, независимо от его природы. Законы Фарадея – основные законы электролиза, согласно которых, количество вещества, выделившегося при электролизе, прямо пропорционально его химическому эквиваленту и количеству прошедшего электричества. Уравнение Нернста Е0 – равновесный стандартный потенциал. 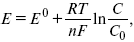 где С0 – стандартная концентрация раствора; С – любая концентрация в нестандартных условиях, С = С0 x Е = Е0 , т. е. в стандартных условиях С = C0 = 1 моль. Для окислительных веществ 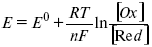 1-й закон Фика: 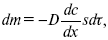 где dc/dx – градиент концентрации; s – площадь, через которую происходит диффузия. ? – коэффициент диффузии cм2 x c-1, показывает число частиц, продиффундировавших за 1 с через поперечное сечение раствора площадью 1 см2, dt – время диффузии, dm – число продиффундировавших частиц. 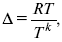 где Тк – коэффициент внутреннего трения; D – коэффициент диффузии. Первый закон Фика относится к процессу стационарной диффузии, сходен с закономерностями переноса тепла из электричества. 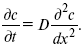 Если диффузионный поток не изменяется с течением времени, это называется стационарной диффузией. Диффузия – самопроизвольно протекающий в системе процесс выравнивания концентрации молекул, ионов, частиц под влиянием теплового хаотического движения. Основное уравнение электрохимической кинетики ik = ia = i0, где i0 – ток обмена,  (окислительно-восстановительные реакции). При катодной поляризации на электроде через систему протекает ik преимущественно, если поляризация не слишком велика, то суммарная скорость процесса равна: i = ik – ia, для реакции (1) катодные и анодные токи будут равны: 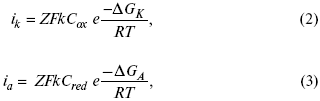 где Z – количество электронов, участвующих в реакции; F – число Фарадея; к – const скорости; Сox, Cred – концентрация окислительной и восстановленной форм реагентов; ?GK – энергия активации катодного процесса; ?GA – энергия активации анодного процесса. Энергия активации зависит от величины накладываемого потенциала, в то же самое время эта энергия распределяется между прямой и обратной реакцией в соответствии с коэффициентом переноса – а, т. е. ? = ?пр – ?об. Коэффициент переноса ?– доля энергии электрического поля в ДЭС, которая приходится на прямую и обратную реакции. ?– коэффициент переноса для катодной реакции; (1 – ?) – для анодного процесса (коэффициент переноса). ?Gk = ZFE ?, (4) ?GA = ZFE(1 – ?) (5) С учетом уравнений (4), (5) уравнения (2), (3) примут следующий вид: 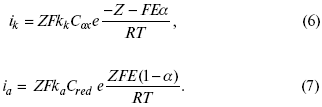 Различие знаков у электрона объясняется тем, что катодная поляризация («–») ускоряет прямую реакцию и замедляет обратную реакцию. 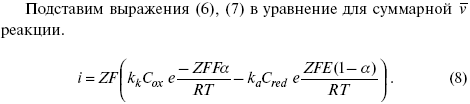 Введем в уравнение (8) плотность тока обмена – i0. 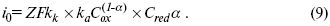 Вместо потенциала введем перенапряжение: 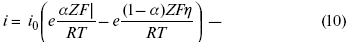 полное уравнение поляризационной кривой. Вывод из уравнения (10): 1) при равновесном потенциале, когда ток равен нулю, уравнение (10) преобразуется в уравнение Нернста: 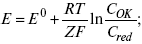 2) при малых величинах ?: 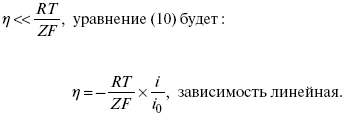 При сдвижении потенциала от равновесного (59 mВ); 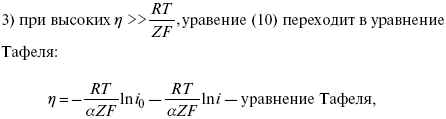 ? = a + b ln i– уравнение Тафеля в простом виде при замедлении стадии переноса заряда. Величина i0 (тока обмена) и ?(коэффициента переноса) – основные кинетические параметры стадии переноса заряда (q). Они могут быть определены из экспериментальных измерений, для этого на исследуемом электроде снимают зависимость ?– i или Ei – i. Поляризационная кривая судит о коррозионной стойкости металлов. Перестраиваем поляризационную кривую в координаты: 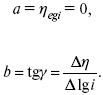 Определяем const а и bв уравнении Тафеля, определяем b: Из коэффициента bнайдем а, после подставим в а и найдем i0. Перенапряжение Н2 (водорода). Источник выделения Н2 – Н2SO4 >Н++ НSO4– Источник выделения Н2 – Н2О > Н++ ОН-. В рН < 7 Н2 выделяется по реакции. 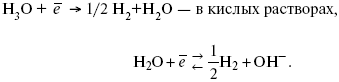 Н2 всегда выделяется в потенциалах более отрицательных, чем равновесный потенциал, то есть с перенапряжением. Суммарный процесс выделения водорода состоит из следующих стадий: 1) доставка к поверхности катода реагирующих частиц Н3О+; 2) разряд Н3О+ с образованием Надс 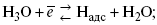 3) удаление выделяющегося Надс с поверхности электрода может происходить тремя путями: а) каталитическая рекомбинация 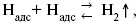 где Кat – материал катода; б) электрохимическая десорбция – удаление Н2 происходит на уже адсорбированных атомах 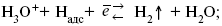 в) эмиссия включает две стадии: 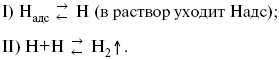 Для Pt замедлена стадия а), для других металлов (Hg, Pb) – стадия разряда, Н+ – самый подвижный. 3. Кинетические особенности электроосаждения металлов и сплавов Процесс электроосаждения металлов, сплавов протекает через последовательность стадий: диффузия катионов металлов к поверхности электрода из объема раствора, вхождение катионов в ДЭС (двойной электрический слой), потери сольватной оболочки, переход катионов в состояние адсорбции атома, полный перенос заряда с поверхности электрода на разряжающийся ион или адсорбированного атома (ад. атома) и образование зародышей металлов, рост зародышей и заполнение поверхности новой фазы в виде сплошного слоя, рост слоя осадка в толщину. Процесс электровыделения не зависит от состояния поверхности электрода, в частности, большое влияние на ?(перенапряжение) процесса оказывает концентрация вакансий на поверхности электрода. Кристаллическая решетка каждого металла содержит определенное количество равновесных вакансий (свободных незанятых узлов в кристаллической решетке). Наличие таких пустот в структуре поверхностного слоя облегчает образование ад.атомов, так как в местах вакансий имеет место более сильное энергетическое воздействие кристаллической решетки на образующиеся атомы новой фазы. После заполнения этих активных мест начинается рост зародышей, т. е. образование скоплений атомов, которые постепенно заполняют всю поверхность. С другой стороны, скорость реакции электровыделения металлов зависит от состояния катионов этого металла в растворе. В растворе катионы находятся в сольватированном виде или в виде комплексов. Разрушение сольватной оболочки происходит на границе плотного слоя Гельмгольца с диффузной частью ДЭС. Таким образом, реакции разряда, протекающие в плотном слое Гельмгольца, энергетически возможны только в том случае, если ионы металла преодолевают потенциальный барьер. Высота потенциального барьера, т. е. величина энергии, которую ионам в растворе нужно преодолеть, чтобы попасть из раствора в плотный слой Гельмгольца, может быть различной, и определяется она природой растворителя, лигандов, прочностью связей в комплексах. Пример: 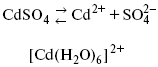 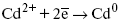 (заряд комплекса не меняется, так как молекула нейтральна). Сама стадия переноса зарядов также протекает стадийно 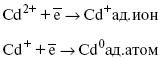 Анионные комплексы наиболее прочные, и последняя стадия состоит из процесса распада до свободного иона на поверхности электрода в слое Гельмгольца. Это обусловлено тем, что анионы, обладающие высокой поверхностной активностью, связываются с поверхностью электрода и оказывают влияние на распределение заряда в ДЭС. Итак, помимо диффузии в объеме раствора, диффузии ад. ионов, ад. атомов по поверхности, стадий переноса заряда, образования зародышей и роста зародышей в сплошной слой (стадия кристаллизации), на скорость реакции могут оказывать влияние также реакции разложения комплексов в растворе, гомогенная химическая стадия, предшествующая стадии разрядов, и гетерогенная химическая стадия на поверхности электродов. Скорость реакции определяется концентрацией потенциал-определяющих частиц в растворе; концентрация потенциал-определяющих частиц зависит от состояния ионов. Состояние ионов в растворе определяется энергией взаимодействия с молекулами растворителя и лигандами. Потенциал электрода определяется активностью ионов раствора. В случае твердых металлических электродов активность самого металлического электрода не сказывается на длительности процесса и на величине скорости потенциала электродов (принято считать ? твердой фазы = 1). Если металл растворен в ртути (Hg), то в этом случае i зависит от ? металла фазы в матрице электрода 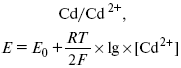 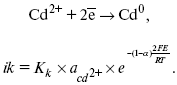 Анодные процессы Ионизация металла (с растворимыми анодами)  С нерастворимыми анодами – реакция выделения кислорода. При повышенных анодных плотностях тока растворимые аноды могут пассивироваться, на поверхности этих анодов образуются фазовые пленки, которые могут быть как токопроводящими, так и непроводящими, в последнем случае ток будет проходить через поры, если пленка не сплошная. Во всех случаях при пассивации анода анодный потенциал будет повышаться, что приведет к изменению анодных реакций и к изменению валентности металла. 4. Влияние природы растворителя на скорость электрохимических реакций Замена одного растворителя на другой скажется на каждой из стадий электрохимического процесса. В первую очередь это отразится на процессах сольватации, ассоциации и комплексообразования в растворах, скажется на стадии диффузии, на скорости процесса разряда ионизации. Изменения в объеме раствора электролита, связанные с процессами сольватации, ассоциации, скажутся на скорости доставки вещества к поверхности электрода и на скорости разряда ионизации. Влияние природы растворителя на стадии разряда ионизации проявляется изменениями константы скорости реакции, коэффициента переноса. Например, при электровосстановлении кадмия из растворов с различными растворителями были получены следующие величины для константы скорости и коэффициента переноса ?– доли энергии (табл. 7). Таблица 7 Получение величины для константы скорости и коэффициента переноса 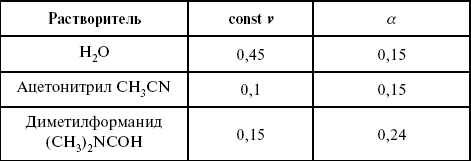 Объяснение изменений кинетических параметров процесса состоит в следующем: 1) изменяется строение ДЭС; 2) изменяется адсорбционная способность разряжающихся частиц; 3) разряжающиеся ионы имеют различную сольватную оболочку. Для объяснения влияния природы растворителя на скорость реакции в объеме раствора была использована теория «Абсолютных скоростей реакции». Основным моментом данной теории является введение понятия «активированный комплекс». Рассмотрим изменение ?G (свободная энергия Гиббса) при замене одного растворителя на другой (рис. 12). 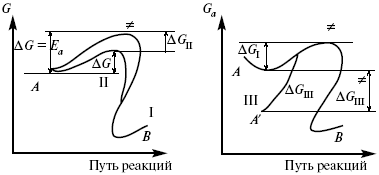 Рис. 12. Энергетические профили реакции. А – исходное энергетическое состояние для реагента, участвовавшего в реакции, В – энергетическое состояние продуктов реакции, ?– активированный комплекс. Для того чтобы прошла реакция в исходное состояние и перешла в состояние активированного комплекса, требуются затраты энергии – Еа. Энергетический профиль, описанный кривой I, соответствует состоянию, когда исходное вещество и активированный комплекс не сольватированы. Энергия, необходимая для превращения вещества А в вещество В для реакции: F = ?GI?. При замене растворителя допустим вариант (а): происходит сольватация активированного комплекса – кривая II. В этом случае наблюдается уменьшение Еа процесса на величину ?GII?. При сольватации исходного реагента (вариант б) – кривая III, происходит увеличение Еа процесса на величину ?GIII?. 5. Электроосмос Электроосмос – перенос жидкости по отношению к граничащей с ней неподвижной твердой поверхностью при приложении ЭДС (электродвижущей силы). Электроосмос возможен только в системах с твердой дисперсной фазой. Электрокинетические процессы происходят в тех случаях, когда одна фаза диспергирована в другой; к их числу относится электрофорез – движение взвешенных твердых частиц внутри жидкости. При наложении электрического поля наблюдается электроосмос – движение жидкости относительно твердого тела. Электрокинетические явления – эффекты, связанные с относительным движением двух фаз под действием электрического поля, а также с возникновением разности потенциалов при относительном смещении двух фаз, на границе между которыми существует ДЭС. Чаще всего электрокинетические явления наблюдаются в диспергированных системах. Электроосмос (электроэндоосмос) – движение жидкостей (или газов) через капилляры, твердые пористые диафрагмы и мембраны, а также через слои очень мелких частиц под действием внешнего электрического поля. Все электрокинетические явления имеют общий механизм и связаны с существованием на границе раздела фаз ДЭС. Под действием внешнего электрического поля, направленного вдоль границ раздела, возникает относительное перемещение противоположно заряженных обкладок ДЭС, что и приводит к относительному движению фаз. С другой стороны, движение одной из фаз по отношению к другой, вызванное механической силой, приводит в относительное движение также обкладки ДЭС и тем самым вызывает появление разности потенциалов в направлении движения фаз. Электроосмос при экспериментальном исследовании обычно осуществляют наложением разности потенциалов на жидкость с двух сторон капилляра или пористой диафрагмы. Поддерживая давление с обеих сторон одинаковым и измеряя в этих условиях количество протекающей в единицу времени жидкости, легко определить скорость электроосмоса. Электроосмос и электрофорез используются при обезвоживании и очистке различных материалов, нанесении на непроводящие материалы покрытий из каучука, отходов кожи и т. п., также при пропитке тканей огнестойкими веществами, определении состава и разделении энзимов, белков, вирусов и других сложных систем. Исследованиями Г. Видемана в 1852 г. было установлено, что количество жидкости, прошедшей через капилляры пористой диафрагмы, пропорционально силе тока и при постоянной силе тока не зависит от площади сечения или толщины диафрагмы. Это явление было названо электроосмосом. Наличие у частиц дисперсных систем электрического заряда открыто в 1808 г. Ф. Ф. Рейсом в МГУ. Он показал, что при наложении разности электрических потенциалов на электроды, опущенные в заполненные водой стеклянные трубки, воткнутые в кусок сырой глины, жидкость в трубке с положительным полюсом мутнела, а в трубке с отрицательным полюсом вода оставалась прозрачной. Это указывало на то, что частицы глины переносятся к положительному полюсу с постоянной скоростью. Эта скорость тем больше, чем выше приложенная разность потенциалов и диэлектрическая проницаемость среды, и тем меньше, чем больше вязкость среды. Перенос частиц в электрическом поле – электрофорез. 6. Электрокапиллярные кривые Изменение состава электролита и изменение компонентов в электролите изменяют электрокапиллярные кривые. Форма электрокапиллярной кривой зависит от состава электролита и концентрации активных компонентов в составе электролита. Зависимость формы электрокапиллярной кривой от состава электролита связана с процессами адсорбции на границе раздела фаз (рис. 13). Присутствие в электролите ПА (поверхностно-активные) анионов приводит к смещению потенциала точки нулевого заряда в область более отрицательного значения и некоторому снижению max электрокапиллярной кривой. В присутствии NaJ, NaCl происходит изменение хода электрокапиллярной кривой. Кривые 2 и 3 – электрокапиллярные кривые, снятые в электролитах, содержащих ПА анионы: J-, Cl–. В области наиболее низких электродных потенциалов все три электрокапиллярные кривые совпадают, так как при указанных потенциалах наблюдается десорбция ПА анионов. В присутствии ПА катионов электрокапиллярные кривые имеют вид: 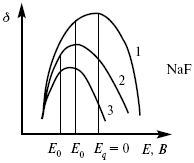 Рис. 13. Кривые 2, 3 в присутствии ПА катионов. Наличие в элементе ПА органических веществ приводит к снижению max электрокапиллярной кривой (рис. 14). 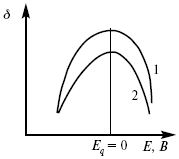 Рис. 14. Кривая 2 – с добавками ПАВ (поверхностно-авктивныхвеществ). Молекулярный тип – не дипольные молекулы 2 – с добавками ПА органическими. Электрокапиллярная кривая – исходная кривая, дифференцируя ее, определяем емкость ДЭС. 7. Электрохимическое перенапряжение (перенапряжение переноса заряда) 1. Вывод уравнения полной поляризационной кривой. 2. Перенапряжение при выделении Н2. 3. Перенапряжение при выделении О2. Если на электроде замедлена стадия присоединения или отдачи электронов, то возникающее перенапряжение – перенапряжение переноса заряда (перенапряжение перехода – электрохимическое перенапряжение). Теория разряжения для реакции выделения Н2 на катоде: 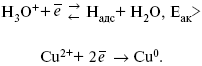 Стадия переноса электрона из-за построения новой кристаллической решетки затруднена. 8. Факторы, влияющие на перенапряжение водорода. Перенапряжение кислорода Факторы, влияющие на ?Н2: 1) ?тока (плотность тока). Зависимость от плотности тока описывается уравнением Тафеля; 2) природа материала катода – ряд по возрастанию ?, ?– перенапряжение. В уравнении Тафеля const a характеризует зависимость ?от природы материала катода, а константа b отражает зависимость от плотности тока. В классическом варианте b – 0,12В, а – меняется в широких пределах, из-за разных металлов и разных катодных взаимодействий с Н2. а – 0,01…1,0 В, чем больше а, тем больше? Н2. Большим ? Н2 обладают: Hg, Pb, Zn, низким ?Н2 – Pt, средним ?Н2 – Fe, Co, Ni; 3) состав раствора. Наибольшее ?в рН = 7, а в рН < 7 ? меньше. В растворе могут быть ПАВ, они влияют на величину?, включаются в плотную часть ДЭС. Уравнение ?в присутствии ПАВ: 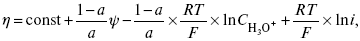 где ?– потенциал плотной части ДЭС; 4) температура. С ростом температуры ?уменьшается. Перенапряжение кислорода Кислород выделяется на аноде при потенциалах более положительных, чем равновесный. 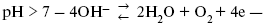 в щелочном растворе, 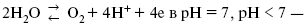 в нейтральном, кислом растворе. Перенапряжение О2 зависит от ?т (плотность тока), в соответствии с уравнением Тафеля. В ряду металлов, расположенных по мере возрастания перенапряжения Н2, перенапряжение О2, наоборот, уменьшается. При увеличении температуры ?О2 снижается. Когда на металле выделяется кислород, то он сразу же окисляет металл, и поэтому дальнейшее выделение кислорода уже проходит на окисленной поверхности. |
|
||
|
Главная | В избранное | Наш E-MAIL | Добавить материал | Нашёл ошибку | Вверх |
||||
|
|
||||